聯(lián)系方式 | 手機(jī)瀏覽 | 收藏該頁(yè) | 網(wǎng)站首頁(yè) 歡迎光臨賦悅科技(杭州)有限責(zé)任公司

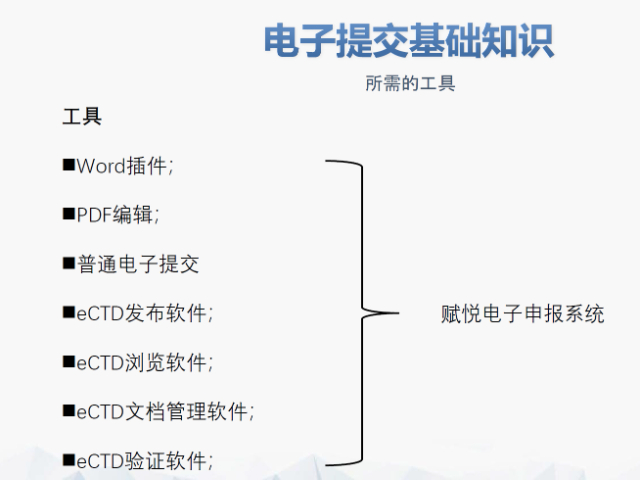
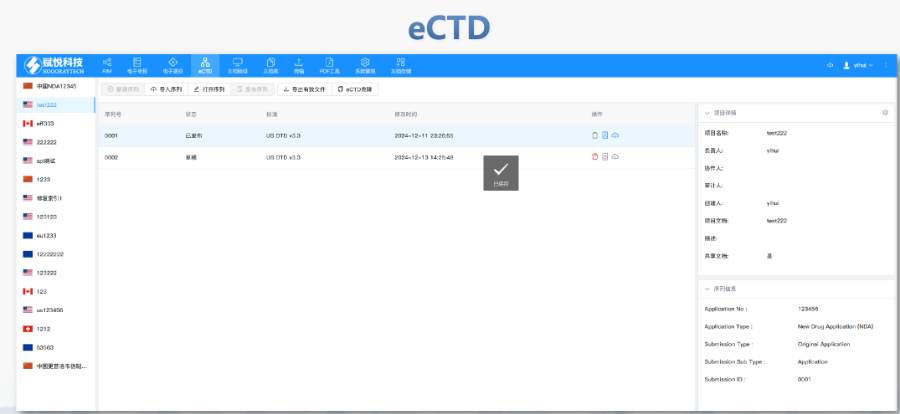
賦悅科技(杭州)有限責(zé)任公司 eCTD藥品申報(bào)系統(tǒng)|普通電子申報(bào)系統(tǒng)|eCTD注冊(cè)外包|全球藥品注冊(cè)咨詢(xún)
13429806270